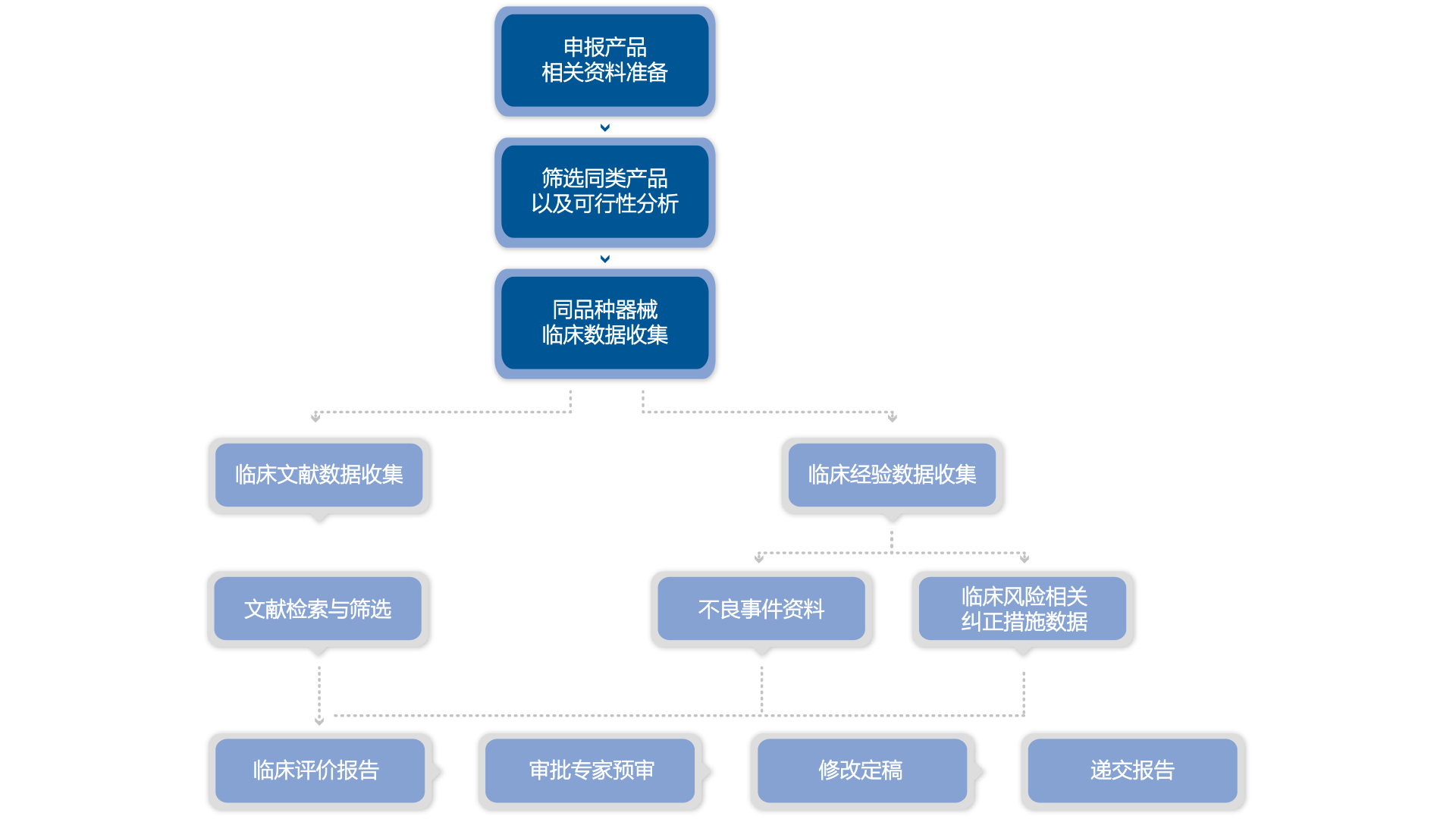
2015年5月NMPA发布《医疗器械临床评价技术指导原则》,自此二、三类医疗器械在NMPA申请注册时,若产品未列于或不符合《免于进行临床试验目录》的条件,则对于上市前的临床审批要求为两条途径:1.提供临床评价资料,也称为“同品种对比报告”、“临床评估报告”、“临床等效评价报告”;2.临床试验。我们需要先判断产品,一个路径走通,再走下一个。以下为适用临床等效评价报告路径的产品情形:
1.进口产品国产化;
2.没及时延续注册,导致需重新注册的产品;
3.产品不变但由于生产商等其他信息的变化造成需换证的产品;
4.已有产品上市,同一企业的后续研发产品(企业有已上市的前代产品或类似产品);
5.已有产品上市,同一集团/企业跨地区办厂的产品;
6. 企业无已上市的前代产品或类似产品,但可以得到类似产品的授权及技术资料;
7. 企业无已上市的前代产品,但产品自身的特点、风险程度和临床安全有效性验证方式适合临床评价(CER)途径。
相比临床试验结果的不可预知性,临床等效评价报告是建立在对已在中国境内上市的同类产品的临床文献资料、临床经验数据、不良事件数据等进行收集分析,判断该产品是否满足使用要求或适用范围的确认过程,风险小且时间大幅缩减。2015年新法规执行后,迈迪思创迅速组建临床等效评价团队,成员均为相关领域硕士及以上学历,且具有5年以上临床等效评价报告编写经验的专业人才。截止目前为止,我们已参与及完成近50份临床评价报告,已取到注册证的产品包括呼吸机、强脉冲光治疗仪、口腔X射线体层摄影设备、电极、一次性麻醉穿刺包、口腔液体敷料、防血栓脉冲贴等,已通过NMPA审评的包括种植体、种植体基台、球囊扩张导管、导管鞘套件、半导体激光脱毛机等。